The entire family of tumor protein p53 (TP53) enhances functions such as apoptosis and autophagy in normal cellular functioning. TP53 is a tumor repressor gene that is often inactivated in human cancers. Reactivating p53 has proven difficult to achieve therapeutically, however. Researchers at MD Anderson Cancer Center are investigating other members of the p53 pathway in order to elucidate new therapeutic options to suppress p53-deficient tumor growth.
ΔN isoforms of two members of the p53 family, p63 and p73, are usually overexpressed in cancers and these isoforms (which lack the acidic transactivation domain) act on p53 in a dominant-negative fashion, blocking its tumor repression abilities [1-6]. Using transfected human cancer cell lines, researchers were able to investigate the roles ΔNp63 and ΔNp73 play in promoting tumor survival. Deleting these ΔN isoforms of p63 and p73 produced tumor regression via the upregulation of IAPP, an amylin-encoding gene that is secreted with insulin. A synthetic analogue of amylin, pramlintide, has proven successful in blocking tumor survival, so this IAPP pathway was analyzed as a new option for treating p53-deficient cancers [7]. Researchers uncovered that TAp63 and TAp73 (isoforms with acidic transactivation domains) transcriptionally regulate IAPP, which in turn blocks glycolysis in the tumor cells, thereby decreasing tumor cell survival [8].
Using Nexcelom’s Celigo, researchers investigated the downstream effects of IAPP, which demonstrated increased rates of apoptosis and the production of reactive oxygen species (ROS) through inhibition of glycolysis [9, 10].
Materials and Methods
*for information regarding the transfection of H1299 cells with ΔNp63, ΔNp73, or IAPP, or the applications of siRNA, please see Venkatanarayan, et al. (2014) “IAPP-driven metabolic reprogramming induces regression of p53 tumours in vivo.” Nature. doi:10.1038/nature13910; http://www.nature.com/nature/journal/vaop/ncurrent/nature13910/metrics
Pramlintide Treatment
Twelve hours after plating, cells were treated with 10μg/mL pramlintide acetate (AMYLIN Pharmaceuticals) or placebo for 48 hours.
Apoptosis Assay
Cells were plated in 96-well plates. After 12 hours, cells were washed with annexin-binding buffer and a mixture of annexin V-Alexa Fluor 488 propidium iodide (PI) and Hoechst 33342. All images were analyzed for percent apoptosis by the Celigo.
ROS Assay
Cells were plated in 96-well plates. After 12 hours, the cells were incubated with CellROX Deep Red Reagent and Hoechst 33342 for 45 minutes. All images were analyzed for percent ROS by the Celigo.
NAC Treatment
Twelve hours after plating, cells were treated with NAC 2mM final concentration for a period of 1 hour.
Results
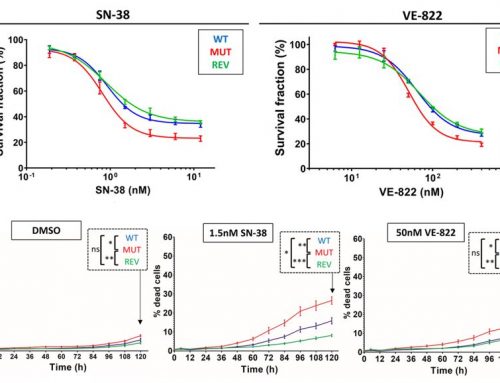
- In human lung adenocarcinoma cells (H1299), the rates of ROS and apoptosis, the latter measured by annexin V/PI staining, increased in cells expressing IAPP or after treatment with pramlintide. In comparison, p53-deficient cells containing ΔNp63 or ΔNp73 showed no reduction in ROS or apoptosis.
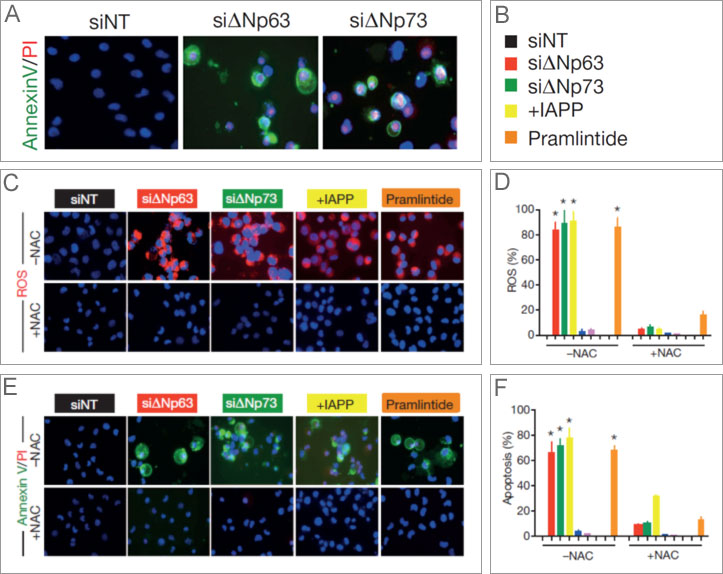
Figure 1a-f. IAPP induces ROS and apoptosis in H1299 cells. Both the knock down of ΔNp63 or ΔNp73 (through siRNA), the expression of IAPP, or treatment with pramlintide increased ROS (c and d) and induced apoptosis (a-top, e, and f). (The addition of N-acetyl-L-cysteine [NAC+, bottom or NAC-, top] blocks ROS and demonstrated that inhibiting ROS in those cells also blocked apoptosis.)
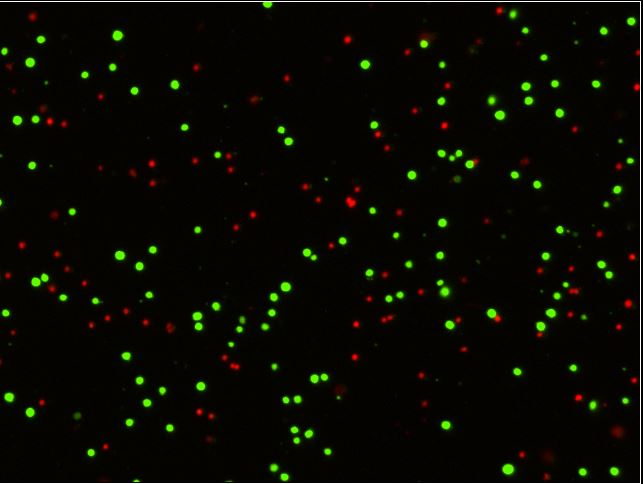
- When media from H1299 cells transfected with ΔNp63 or ΔNp73 and containing secreted IAPP was added to naïve H1299, the cells demonstrated higher levels of apoptosis, as measured by annexin V/PI staining, as well as increased ROS.
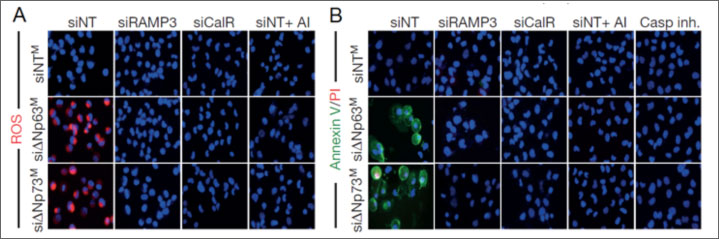
Figure 2a and b. Secreted IAPP from ΔNp63 or ΔNp73 transfected H1299 cells was capable of inhibiting glycolysis in naïve H1299 cells, thereby increasing the percentage of ROS (a) and apoptosis (b) positive cells.
Conclusion
- Exploiting the IAPP pathway may yield novel methods of treating p53-deficient cancers.
*Note: the paper outlining these experiments in more detail may be found here [http://www.nature.com/nature/journal/vaop/ncurrent/nature13910/metrics]
References
- Flores, E.R., et al., Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell, 2005. 7(4): p. 363-73.
- Su, X., et al., TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature, 2010. 467(7318): p. 986-90.
- Su, X., D. Chakravarti, and E.R. Flores, p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer, 2013. 13(2): p. 136-43.
- Tomasini, R., et al., TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev, 2008. 22(19): p. 2677-91.
- Yang, A., et al., p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell, 1998. 2(3): p. 305-16.
- Su, X., et al., TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab, 2012. 16(4): p. 511-25.
- Edelman, S., H. Maier, and K. Wilhelm, Pramlintide in the treatment of diabetes mellitus. BioDrugs, 2008. 22(6): p. 375-86.
- Castle, A.L., et al., Amylin-mediated inhibition of insulin-stimulated glucose transport in skeletal muscle. Am J Physiol, 1998. 275(3 Pt 1): p. E531-6.
- Mattson, M.P. and Y. Goodman, Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res, 1995. 676(1): p. 219-24.
- Schubert, D., et al., Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci U S A, 1995. 92(6): p. 1989-93.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment